So funktioniert UV-Spektrometrie
Ob im Biochemie-Praktikum, im Qualitätslabor eines Pharmaherstellers oder bei der Gewässerüberwachung: Die UV-Spektrometrie ist aus der modernen Analytik nicht wegzudenken. Während die NIR-Spektrometrie mit Molekülschwingungen arbeitet, setzt die UV-Spektrometrie eine Ebene tiefer an: bei den Elektronen selbst. Was auf dieser Ebene physikalisch passiert, schauen wir uns im Folgenden genauer an.
Elektronen und ihre Energieniveaus
Wer im Chemieunterricht aufgepasst hat, erinnert sich: Elektronen umkreisen den Atomkern auf Orbitalen. Was im Unterricht oft unter den Tisch fällt: Diese Orbitale sind keine x-beliebigen Bahnen, sondern präzise definierte Energieniveaus. Und genau hier beginnt die Geschichte der UV-Spektrometrie.
Sobald Atome eine Bindung eingehen, überlappen ihre Atomorbitale und bilden Molekülorbitale. Anders als bei isolierten Atomen gibt es nun drei Sorten, die sich energetisch unterscheiden.
Bindende Orbitale entstehen, wenn sich die Wellenfunktionen der Atomorbitale konstruktiv überlagern, bildlich gesprochen: Die Elektronenwolken addieren sich zwischen den Atomkernen. Das Ergebnis ist ein Orbital mit niedrigerer Energie als die beiden ursprünglichen Atomorbitale. Elektronen, die sich hier aufhalten, stabilisieren das Molekül. Die σ-Orbitale (Sigma, [ˈzɪɡma]) einer Einfachbindung oder die π-Orbitale (Pi, [piː]) einer Doppelbindung gehören in diese Kategorie.
Antibindende Orbitale (gekennzeichnet mit einem Stern: σ* [Sigma-Stern], π* [Pi-Stern]) sind das Gegenstück. Sie entstehen durch destruktive Überlagerung der Wellenfunktionen: Zwischen den Atomkernen bildet sich eine Knotenebene ohne Elektronendichte. Weil die Elektronen die Kerne nicht mehr abschirmen, stoßen sich die positiv geladenen Kerne stärker ab. Das Orbital ist energetisch angehoben und im Grundzustand unbesetzt. Antibindende Orbitale sind die „Ziellinie" eines elektronischen Übergangs.
Nichtbindende Orbitale, kurz n-Orbitale (gesprochen: Enn, [ɛn]), sind ein Sonderfall. Sie gehen nicht aus der Überlappung von Atomorbitalen hervor, sondern existieren als atomare Restorbitale, die an der Bindung gar nicht beteiligt sind. Das klassische Beispiel sind die freien Elektronenpaare des Sauerstoffs in einer Carbonylgruppe (C=O): Der Sauerstoff bringt zwei Elektronenpaare mit, die an keiner Bindung mitwirken, aber für die UV-Spektrometrie eine große Rolle spielen, dazu gleich mehr.
Im Grundzustand besetzen die Elektronen die energetisch niedrigsten verfügbaren Orbitale. Das höchste besetzte Orbital nennt man HOMO (Highest Occupied Molecular Orbital), das niedrigste unbesetzte LUMO (Lowest Unoccupied Molecular Orbital).
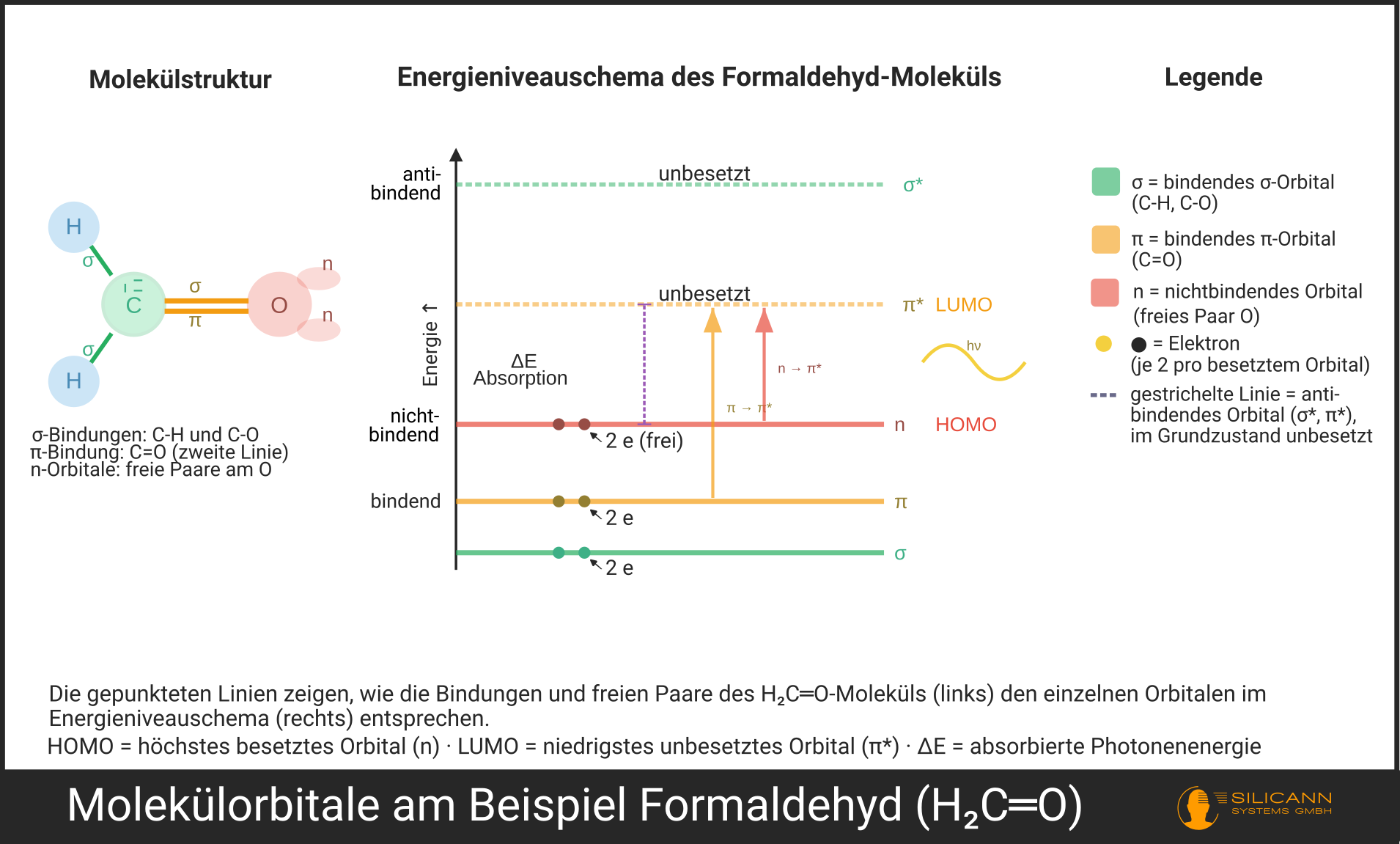
Der Energieabstand zwischen HOMO und LUMO ist der entscheidende Faktor für die UV-Spektrometrie. Denn genau dieser Abstand bestimmt, welche Wellenlänge ein Molekül absorbieren kann.
Wie Moleküle UV-Strahlung absorbieren
Trifft elektromagnetische Strahlung auf ein Molekül, so kann ein Elektron die Energie eines Photons aufnehmen und aus einem besetzten in ein unbesetztes Orbital springen: aus dem Grundzustand in einen angeregten Zustand. Das ist der zentrale Vorgang der UV-Spektrometrie.
Die Bedingung dafür: Die Energie des Photons muss exakt dem Energieabstand zwischen den beiden Orbitalen entsprechen. Liefert das Photon zu wenig oder zu viel, passiert: nichts. Das Molekül bleibt im Grundzustand. Es gelten hier dieselben strengen Quantenregeln wie bei den Molekülschwingungen der Infrarot-Spektrometrie.
Die Energie eines Photons hängt von seiner Wellenlänge ab:
\(E = \frac{h \cdot c}{\lambda}\)
Die Faustregel: je kürzer die Wellenlänge, desto größer die Energie des Photons. UV-Strahlung (200-400 nm) ist kurzwelliger als sichtbares Licht und trägt damit genug Energie, um Elektronen zwischen Orbitalen springen zu lassen. Sichtbares Licht und erst recht Infrarot-Strahlung reichen dafür energetisch nicht aus; hier dominieren die Molekülschwingungen, wie im Artikel zu Grund- und Oberschwingungen beschrieben.
Die Quantenebene
Wer sein erstes UV-Spektrum sieht und statt eines glatten Kurvenverlaufs markante Peaks erblickt, stellt unweigerlich die Frage: Warum absorbiert das Molekül nicht einfach alle Wellenlängen ein bisschen?
Die Antwort ist ein quantenmechanischer Effekt, der den Bauplan des Universums gleich mitliefert. Elektronen können eben nicht nach dem Gießkannenprinzip jede beliebige Energiemenge aufnehmen. Ihre Orbitale lassen sich nur in diskreten Sprüngen wechseln. Eine Erkenntnis, die Nils Bohr Anfang des 20. Jahrhunderts am Wasserstoffatom erstmals mathematisch fasste.
Photonen, deren Energie nicht exakt einem erlaubten Übergang entspricht, passieren das Molekül einfach ungehindert. Das Ergebnis ist ein Spektrum, in dem nur bei ganz bestimmten Wellenlängen Absorption stattfindet.
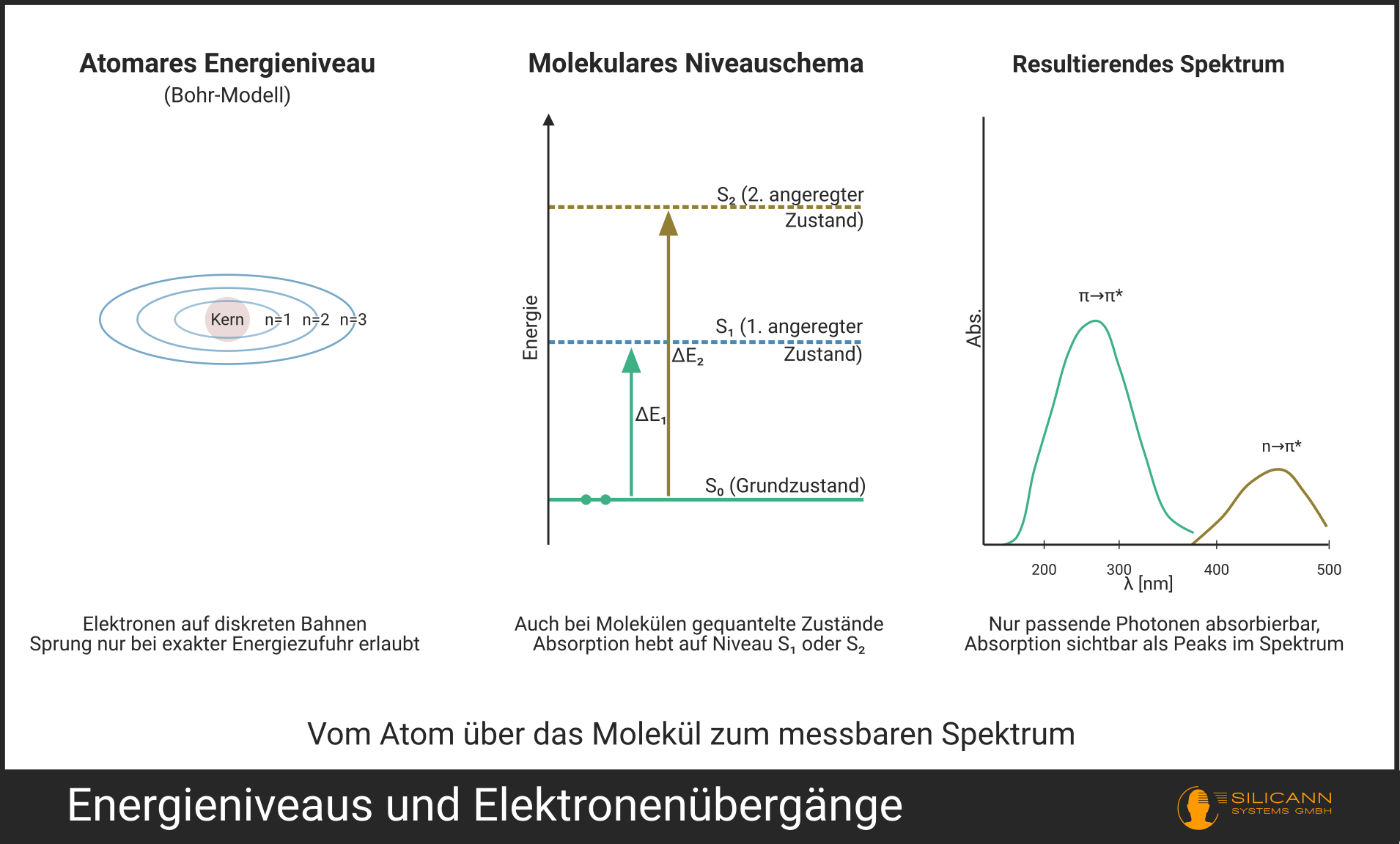
Ein genauerer Blick ins reale UV-Spektrum offenbart allerdings keine Ansammlung messerscharfer Linien, wie man sie von atomaren Spektren kennt. Stattdessen zeigen sich breitere Banden. Woran liegt das? Jeder elektronische Übergang wird von einer Vielzahl gleichzeitiger Schwingungs- und Rotationsanregungen des Moleküls begleitet. Die einzelnen, minimal gegeneinander verschobenen Übergänge überlagern sich und laufen zu einer breiten Bande zusammen. Was nach einem Nachteil in der Auflösung klingt, ist aus der Sicht der Routineanalytik ein Geschenk: Mit sehr schmalen atomaren Linien wäre im Alltag kaum zu arbeiten. Der Grund: Atomare Peaks sind oft nur Bruchteile eines Nanometers breit. Ein Spektrometer, das solche Feinheiten auflösen kann, wäre zu teuer und empfindlich für die praktische Nutzbarkeit. Schon eine geringfügige Temperaturänderung kann das Gitter minimal verstimmen, bei einer 20 nm breiten Bande fällt das nicht auf, bei einer 0,1 nm breiten Linie verschwindet der Peak jedoch komplett. Für die Konzentrationsbestimmung müsste man zudem exakt das Peakmaximum treffen, während eine breite Bande auch bei leichten Abweichungen der Wellenlänge stabile und reproduzierbare Messwerte liefert. Die molekularen Banden sind also kein Bug, sondern ein Feature: Sie machen die UV-Spektrometrie robust genug für die Prozessüberwachung mit erschwinglicher Hardware.
Die Typen elektronischer Übergänge
Nicht jedes Elektron kann durch UV-Strahlung angeregt werden. Welche Übergänge möglich sind, hängt von der elektronischen Struktur des Moleküls ab. Man unterscheidet vier Haupttypen (der Pfeil →, gesprochen „nach", markiert den Weg des Elektrons vom Ausgangs- zum Zielorbital):
| Übergang | Beschreibung | Typische Wellenlänge | Beispiel |
|---|---|---|---|
| \(\sigma \rightarrow \sigma^*\) | Bindungselektron einer Einfachbindung springt ins antibindende Orbital | < 200 nm (Vakuum-UV) | C-H, C-C-Einfachbindungen |
| \(n \rightarrow \sigma^*\) | Freies Elektronenpaar springt ins antibindende σ-Orbital | 150-250 nm | H₂O, Alkohole, Ether |
| \(\pi \rightarrow \pi^*\) | π-Elektron einer Doppelbindung springt ins antibindende π-Orbital | 200-700 nm | C=C, C=O, Aromaten |
| \(n \rightarrow \pi^*\) | Freies Elektronenpaar springt ins antibindende π-Orbital | 250-600 nm | C=O, N=N, Nitroverbindungen |
\(\sigma \rightarrow \sigma^*\)-Übergänge erfordern die meiste Energie. Sie liegen im fernen UV-Bereich unterhalb von 200 nm, der nur mit Vakuum-Spektrometern zugänglich ist; daher die Bezeichnung „Vakuum-UV“. Für die Routine-UV-Spektrometrie spielen sie kaum eine Rolle.
Die praktisch wichtigsten Übergänge sind \(\pi \rightarrow \pi^*\) und \(n \rightarrow \pi^*\). Sie liegen im messbaren UV-Bereich (200-400 nm) und sind charakteristisch für Moleküle mit Doppelbindungen, also für fast alle organischen Substanzen von Interesse.
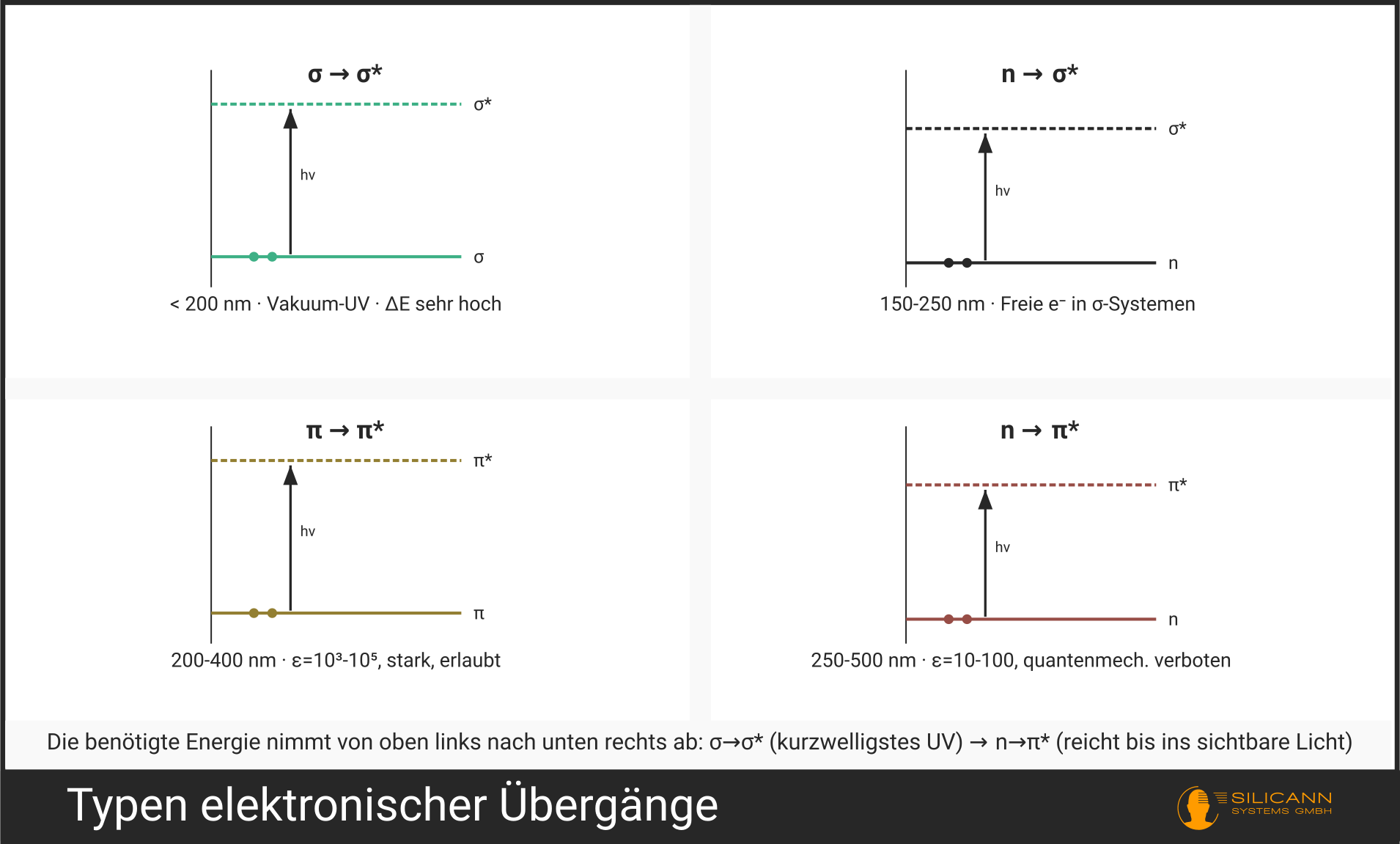
Der \(n \rightarrow \pi^*\)-Übergang ist besonders interessant: Er ist quantenmechanisch „verboten“ (formal von geringer Wahrscheinlichkeit), findet aber dennoch statt, nur mit entsprechend schwächerer Intensität. Das unterscheidet ihn auf den ersten Blick im Spektrum von den erlaubten \(\pi \rightarrow \pi^*\)-Übergängen desselben Moleküls.
Chromophore und Auxochrome
Nicht jedes Molekül ist im UV-Spektrometer sichtbar. Also: Welche sind es? Das Zauberwort heißt Chromophor (wörtlich: „Farbträger“). Der Begriff stammt zwar aus der VIS-Spektroskopie, seine Prinzipien lassen sich aber eins zu eins in den UV-Bereich übertragen.
Ein Chromophor ist eine funktionelle Gruppe, die elektromagnetische Strahlung absorbiert. In der UV-Spektrometrie sind das vor allem Gruppen mit π-Elektronen:
- Einfache Chromophore: Die Carbonylgruppe C=O (π→π* und n→π*), die Nitrogruppe NO₂, die Azogruppe N=N, das Imin C=N.
- Konjugierte Systeme: Mehrere Doppelbindungen im Wechsel mit Einfachbindungen (C=C-C=C-C=C …). Je länger das konjugierte System, desto niedriger die benötigte Anregungsenergie und desto weiter wandert die Absorption in Richtung längerer Wellenlängen. Als grobe Faustregel gilt: Ab etwa 8 konjugierten Doppelbindungen verschiebt sich die Absorption oft so weit, dass sie den sichtbaren Bereich erreicht – das Molekül erscheint farbig. Die Übergänge sind hier jedoch fließend.
- Aromaten: Benzol und seine Derivate haben charakteristische π→π*-Absorptionen, die mit zunehmender Substitution komplexer werden.
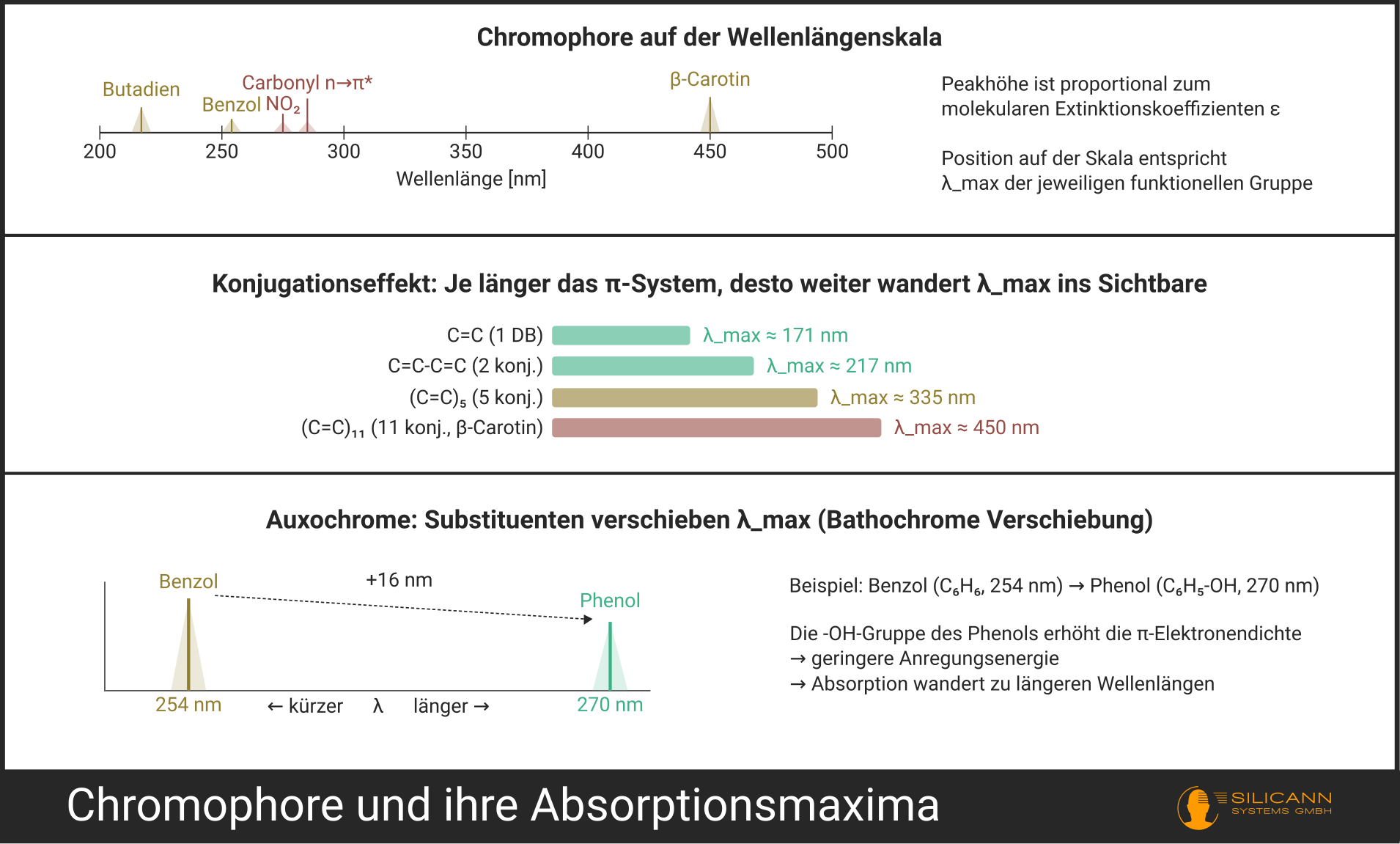
Auxochrome sind Substituenten mit freien Elektronenpaaren (wie -OH, -NH₂, -OCH₃, Halogene), die selbst nicht im UV absorbieren, aber die Absorption benachbarter Chromophore beeinflussen. Sie verschieben das Absorptionsmaximum und verändern die Intensität. Chemisch gesprochen: Sie erhöhen die Elektronendichte im konjugierten System und verringern dadurch die Anregungsenergie.
Lösungsmitteleffekte
Viele unterschätzen die Rolle des Lösungsmittels. Dabei ist es alles andere als ein passiver Zuschauer. Es verändert sowohl die genaue Position als auch die Form der Absorptionsbanden, ein Effekt, den die Fachwelt Solvatochromie nennt.
Die wichtigsten Effekte:
- Bathochrome Verschiebung (Rotverschiebung): Das Absorptionsmaximum wandert zu längeren Wellenlängen. Das passiert besonders bei \(\pi \rightarrow \pi^*\)-Übergängen in polaren Lösungsmitteln, weil der angeregte Zustand stärker stabilisiert wird als der Grundzustand.
- Hypsochrome Verschiebung (Blauverschiebung): Das Maximum wandert zu kürzeren Wellenlängen. Typisch für \(n \rightarrow \pi^*\)-Übergänge in protischen Lösungsmitteln (wie Wasser oder Ethanol), weil das freie Elektronenpaar durch Wasserstoffbrückenbindungen im Grundzustand stabilisiert wird.
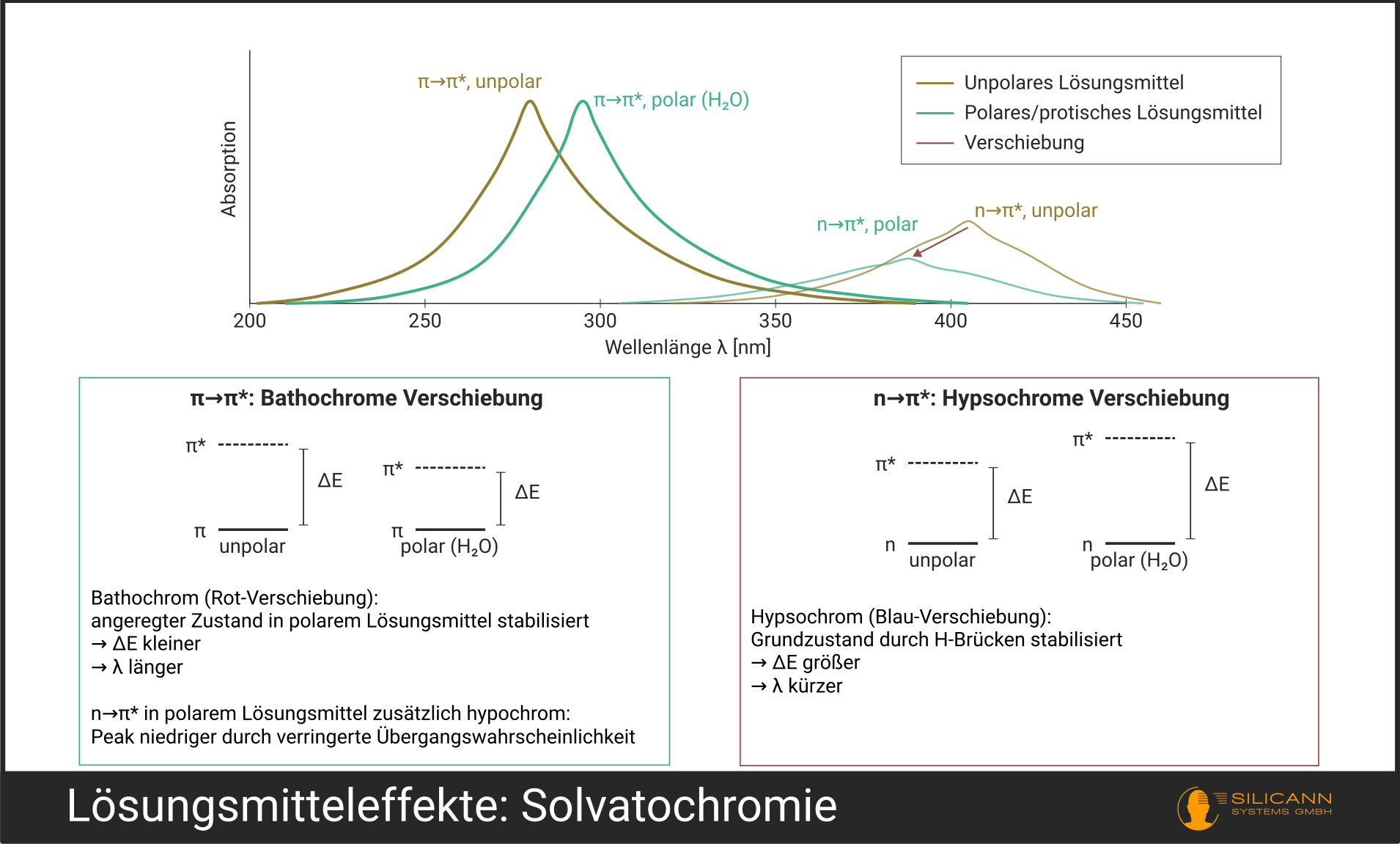
Daneben gibt es hyperchrome (intensivere Absorption) und hypochrome Effekte (schwächere Absorption), die bei veränderten Übergangswahrscheinlichkeiten durch die Lösungsmittelumgebung auftreten.
Kurzum: Ohne Angabe des Lösungsmittels ist ein UV-Spektrum streng genommen unvollständig. In der Praxis wird deshalb die Messung gegen das reine Lösungsmittel referenziert (das sogenannte „Nullabgleichen“), sodass die Eigenabsorption des Lösungsmittels aus dem Ergebnis herausfällt.
UV im Vergleich zu VIS und NIR
Die verschiedenen Bereiche der optischen Spektroskopie unterscheiden sich nicht nur in der Wellenlänge; sie beruhen auf fundamental verschiedenen physikalischen Mechanismen.
| Bereich | Wellenlänge | Physikalischer Vorgang | Typische Information |
|---|---|---|---|
| UV | 200-400 nm | Elektronenübergänge | Chromophore, Konjugation, Aromaten |
| VIS | 400-780 nm | Elektronenübergänge | Farbe, Metallkomplexe, Konjugate |
| NIR | 780-2500 nm | Molekülschwingungen (Oberschwingungen) | OH, CH, NH, funktionelle Gruppen |
| MIR | 2500-25.000 nm | Molekülschwingungen (Grundschwingungen) | Detaillierte Strukturaufklärung |
In der UV-Spektrometrie blickt man auf die elektronische Struktur eines Moleküls, auf das π-System und den Grad der Konjugation. In der NIR-Spektrometrie steht dagegen die mechanische Schwingungsstruktur im Vordergrund.
Beide Methoden ergänzen sich. UV-Messungen sind aufgrund ihrer sehr hohen molaren Extinktionskoeffizienten äußerst empfindlich und eignen sich hervorragend für die Spurenanalytik spezifischer Gruppen. NIR liefert hingegen ein ganzheitlicheres Bild der Molekülstruktur und ist unschlagbar in der quantitativen Analyse komplexer Gemische.

Ein Vorteil der UV-Spektrometrie: Die molaren Extinktionskoeffizienten elektronischer Übergänge (\(10^3\)-\(10^5\) L·mol⁻¹·cm⁻¹) sind um Größenordnungen höher als diejenigen von Schwingungsübergängen in der NIR-Spektrometrie. UV-Messungen sind daher in der Regel weit empfindlicher. Das ist der Grund, warum sich die Methode so hervorragend für die quantitative Analyse eignet, oft bereits im Spurenbereich.
Von der Physik zur Messung: Das Lambert-Beersche Gesetz
Bevor wir zur Anwendung kommen, fehlt noch ein wichtiges Puzzleteil: Wie wird aus einem springenden Elektron ein konkreter Konzentrationswert? Das Spektrometer misst zunächst nur, wie viel Licht durch die Probe hindurchkommt – die Transmission (\(T\)). Daraus berechnet es logarithmisch die Absorption (\(A\)). Im Laboralltag und in Arzneibüchern wird hierfür oft synonym der Begriff „Extinktion“ verwendet, da dieser Begriff zusätzlich Lichtstreuung an Partikeln mit einschließt.
Der Zusammenhang zwischen dieser gemessenen Lichtabschwächung und der Konzentration ist das Herzstück der quantitativen Spektrometrie: das Lambert-Beersche Gesetz.
\(A = \varepsilon \cdot c \cdot d\)
Die Absorption (\(A\)) ist das Produkt aus dem molaren Extinktionskoeffizienten (\(\varepsilon\)), der Konzentration (\(c\)) und der Schichtdicke der Küvette (\(d\)). Es ist ein herrlich einfaches, lineares Gesetz: Doppelte Konzentration bedeutet doppelte Absorption.
Physikalisch gilt dieses Gesetz für den gesamten Spektralbereich. Wenn die Kalibriergerade im Labor bei sehr hohen Konzentrationen trotzdem manchmal abflacht, liegt das nicht am Gesetz selbst, sondern an den Grenzen der Messrealität: Falschlicht im Gerät, nicht ideal-monochromatisches Licht oder physikalische Wechselwirkungen der Moleküle in zu dichten Lösungen stören dann die Linearität. Bleibt man aber im Bereich verdünnter, klarer Lösungen, ist das Lambert-Beersche Gesetz ein felsenfester Anker.
Anwendungsfelder der UV-Spektrometrie
Es gibt kaum ein analytisches Labor, das ohne UV-Spektrometrie auskommt. Ein Blick auf die wichtigsten Einsatzfelder zeigt, warum:
- Konzentrationsbestimmungen: Die Stärke der Absorption ist proportional zur Konzentration des absorbierenden Stoffs. Damit lassen sich Konzentrationen direkt messen: schnell, zerstörungsfrei und mit einer unkomplizierten linearen Kalibration. Im Gegensatz zur NIR-Spektrometrie, die oft komplexe chemometrische Rechenmodelle erfordert, genügt hier in der Regel eine einfache Verdünnungsreihe als Referenz.
- Reinheitsprüfung und Qualitätskontrolle: Viele Pharmazeutika, Farbstoffe und Lebensmittelzusätze haben charakteristische UV-Spektren. Abweichungen deuten auf Verunreinigungen hin.
- Verfolgung chemischer Reaktionen: Verschwindet ein Edukt-Peak oder entsteht ein Produkt-Peak, lässt sich der Reaktionsfortschritt in Echtzeit verfolgen; Grundlage der Reaktionskinetik.
- Protein- und DNA-Quantifizierung: Proteine absorbieren bei 280 nm (aromatische Aminosäuren), DNA bei 260 nm. Die UV-Spektrometrie ist die Standardmethode zur Konzentrationsbestimmung in Biochemie und Molekularbiologie.
- Gewässeranalytik: Nitrat, Nitrit, gelöste organische Substanzen und viele Schadstoffe absorbieren im UV und lassen sich so direkt im Wasser nachweisen.
Die Hardware dahinter reicht vom klassischen Labor-Spektrometer bis zu kompakten, prozessbegleitenden Inline-Spektrometern, die kontinuierlich im Prozess messen und so die Echtzeit-Überwachung der Produktion ermöglichen.
Dass die UV-Spektrometrie aus diesen Feldern nicht wegzudenken ist, liegt neben ihrer analytischen Stärke auch an einem pragmatischen Grund: Die Technik ist sehr ausgereift. Mit robusten Deuteriumlampen, Quarzglas-Optiken und Silizium-Detektoren steht eine zuverlässige und wirtschaftliche Instrumentierung zur Verfügung. Deshalb spielt die UV-Spektrometrie auch in der automatisierten Prozesskontrolle eine tragende Rolle.
Vielleicht auch interessant
KI-unterstützte Entwicklung für Spektralwerk-Spektrometer
Die Spektralwerk-API-Dokumentation ist nun als MCP-Server verfügbar. Das erleichtert die KI-gestützte Entwicklung insbesondere in sicherheitsorientierten Unternehmen.


